Los síntomas más frecuentes de los tumores cerebrales son por el siguiente orden: el déficit neurológico progresivo, generalmente la aparición de debilidad en extremidades, la cefalea intensa y las crisis epilépticas. Otros signos asociados a los tumores cerebrales son la alteración del lenguaje, la aparición de una demencia rápidamente progresiva, la afectación visual y la alteración de la marcha.
Aparte de los síntomas citados, los tumores cerebrales en niños pueden manifestarse con vómitos, irritabilidad, visión doble, afectación del equilibrio y dificultad para tragar.
Entre las causas de los tumores cerebrales, lo cierto es que la mayor parte de ellos surgen de un modo espontáneo, esto es, no están provocados por un factor genético único. Existen, no obstante, un porcentaje pequeño de casos en los que una alteración genética da lugar a la aparición de síndromes que llevan asociados una alta frecuencia de tumores cerebrales. Se trata de los denominados síndromes neurocutáneos o facomatosis. Estas alteraciones asocian diferentes marcas o estigmas a nivel de la piel como angiomas, lesiones protruyentes, manchas café con leche o hipopigmentadas. Están representados fundamentalmente por la Neurofibromatosis I o enfermedad de von Recklinghausen, Neurofibromatosis II, la Esclerosis tuberosa o enfermedad de Bourneville, la enfermedad de von Hippel Lindau y el síndrome de Sturge Weber. Constituyen en su conjunto causas de tumores cerebrales sobre todo en niños y en personas jóvenes. Otros trastornos genéticos relacionados con el desarrollo de tumores en otras localizaciones como en síndrome de Cowden, Turcot o Li-Fraumeni se asocian también con la aparición de tumores cerebrales. Entre las causas ambientales que provocan tumores cerebrales, las radiaciones ionizantes constituyen la más conocida. De esta manera, aquellas personas que han recibido tratamiento con radioterapia a nivel craneal presentan mayor riesgo de desarrollar tumores cerebrales. Por otro lado, no ha sido demostrada la asociación entre el uso del teléfono móvil y la aparición de tumores cerebrales.
A continuación, se enumeran los tipos más frecuentes de tumores cerebrales en adultos:
–Meningiomas: Se trata del tumor cerebral benigno más frecuente. Su origen se sitúa en una de las capas meningeas que rodean el cerebro, en concreto la aracnoides. En un 90% de los casos los meningiomas corresponden al grado I de la clasificación de la Organización Mundial de la Salud (en adelante OMS) lo que les confiere un comportamiento biológico benigno. Muy raros en niños, son más frecuentes en mujeres aumentando su incidencia conforme avanza la edad. En caso de presentar síntomas, crecimiento progresivo o efecto compresivo sobre las estructuras intracraneales es preciso su tratamiento, generalmente quirúrgico. Los casos menores de 3 cm y con una localización profunda pueden beneficarse de la Radiocirugía. Aquellos casos con un diagnóstico incidental (los encontrados de forma inesperada debido a una prueba de imagen motivada por otra causa) sólo precisarán tratamiento en menos de un tercio de los casos, permaneciendo el resto estables y asintomáticos.
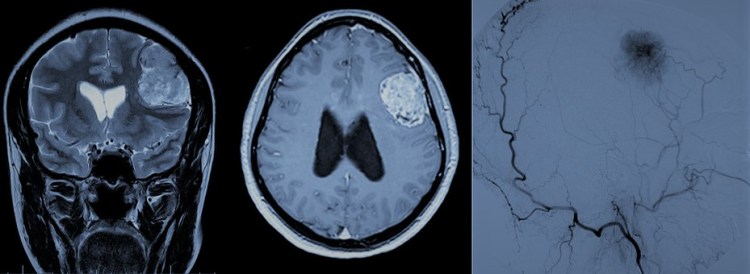
-Gliomas: Son tumores originados de células gliales, esto es, las células que sirven de apoyo estructural y fucional a las neuronas. Ello provoca que sean tumores con mayor capacidad infiltrativa, lo que en general favorece las recurrencias.
A continuación se describen los tipos de gliomas:
-Astrocitomas: Se subdividen a su vez en pilocíticos (grado I de la OMS) que aparecen en niños, difusos (grado II de la OMS), anaplásicos (grado III de la OMS) y glioblastomas (grado IV). Existe asociación entre la edad y el grado de la OMS, de manera que a mayor edad estos tumores suelen presentar un comportamiento más agresivo (mayor grado de la OMS) y con ello mayor tendencia a la recidiva. La última clasificación de los tumores la OMS otorga gran importancia al estado del gen de la Isocitrato Deshidrogenasa (IDH-1 y 2) en estos tumores. En los grado IV el estado del enzima MGMT va a determinar también su grado de agresividad y de respuesta a tratamiento. La base del tratamiento de estos tumores es quirúrgico, estando indicado el tratamiento adyuvante con Quimio y Radioterapia en la mayoría de los casos a partir del grado II de la OMS.

–Oligodendrogliomas: Son tumores derivados de los oligodendrocitos, las células encargadas de formar la mielina en el sistema nervioso central. Pueden pertenecer al grado II o III de la OMS (en el caso de los anaplásicos). Desde el punto de vista genético se definen por la codelección en los cromosomas1p/19q. Su comportamiento clínico y radiológico es muy parecido al de los astrocitomas del correspondiente grado de la OMS, si bien los oligodendrogliomas van a tener un mejor pronóstico. Los aspectos del tratamiento son idénticos a los de los astrocitomas.
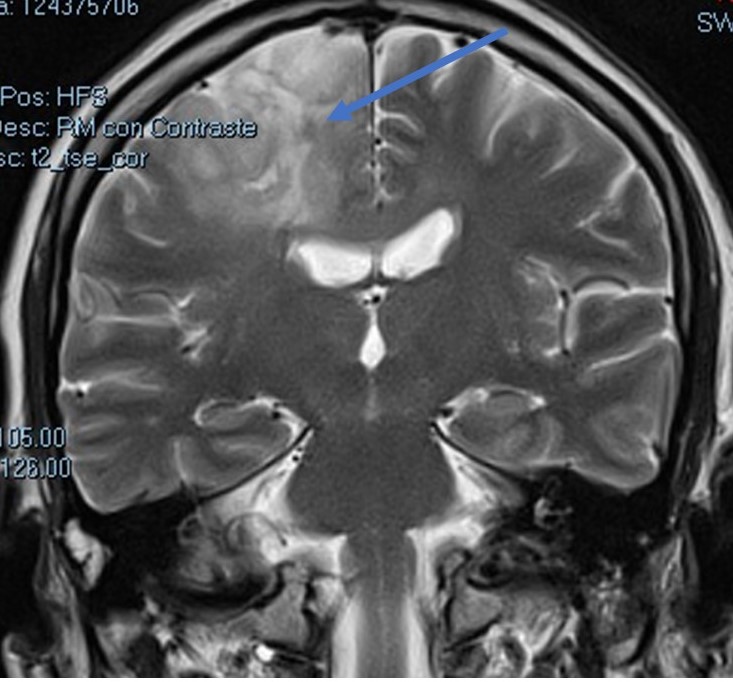
–Ependimomas: En general menos frecuentes que los anteriores, si bien representan uno de los tipos tumorales más frecuentes en niños. Derivan de la célula del epéndimo, aquella que recubre el sistema ventricular. Característicamente se localizan en la fosa posterior, en concreto en el suelo del IV ventrículo, si bien pueden aparecer también en localización supratentorial (en estos últimos la caracterización molecular tiene especial importancia pronóstica de cara a establecer el estado de la fusión RELA). Su contacto con el neuroeje obliga a descartar que presenten extensión a la médula espinal. Su tratamiento es quirúrgico seguido de radioterapia.
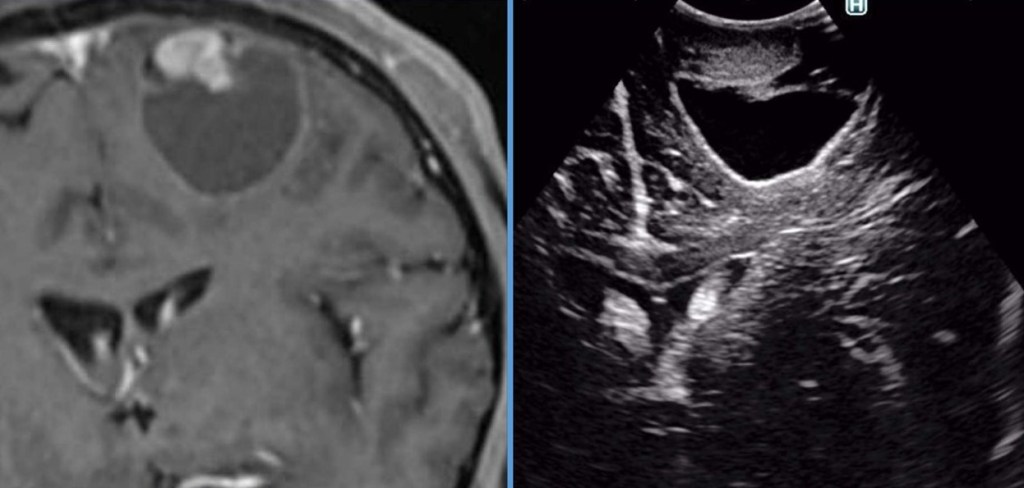
-Schwannomas (Neurinomas): Se trata de tumores originados a partir de las células de Schwann, encargadas de formar la mielina de los nervios ( la capa grasa aislante que los recubre y permite que puedan llevar a cabo la conducción eléctrica de los estímulos nerviosos). A nivel intracraneal la localización más frecuente de los schwannomas tiene lugar en el ángulo pontocerebeloso, donde representan el tumor más frecuente. En esta localización se originan a partir del octavo par craneal (VIII pc), esto es, el nervio vestibulococlear (o estatoacústico) encargado de la audición y el equilibrio. Es, por ello, muy frecuente que la pérdida de audición unilateral sea el síntoma inicial que conduzca al diagnóstico. El tratamiento de elección es la cirugía, si bien aquellos menores de 3 cm puden estabilizarse gracias al tratamiento con Radiocirugía.

-Adenomas hipofisarios: Tienen lugar en la hipófisis, glándula de secreción interna albergada en la silla turca del hueso esfenoides, a nivel de la base del cráneo. Esta glándula se encuentra integrada con el hipotálamo y conjuntamente con éste regula la función de los diferentes órganos endocrinos. Los adenomas de hipófisis se subdividen según su tamaño en microadenomas, menores de un centímetro, y macroadenomas, a partir de un centímetro y según su actividad hormonal en “funcionantes”, aquellos que generan una producción aumentada de hormonas lo que a va a su vez va a generar un síndrome endocrinológico, y “no funcionantes”, aquellos que no generan producción hormonal adicional. Se trata, en general, de tumores frecuentes, sobre todo en el caso de los microadenomas. En todos los casos se debe consultar con un Endocrinólogo de cara a descartar una disfunción hormonal. En el caso de los macroadenomas, se precisa además valoración por un Oftalmólogo pues su localización próxima a los nervios ópticos puede dar lugar a la pérdida de agudeza visual (que lamentablemente a veces constituye el síntoma inicial). Las posibilidades de tratamiento van a estar condicionadas por varios aspectos, fundamentalmente el tamaño del tumor, la afectación o no de la visión y la producción o no de un síndrome debido a la hipersecreción hormonal. El tratamiento médico está especialmente indicado en tumores secretores de Prolactina (Prolactinomas). La cirugía, por su parte está indicada cuando hay afectación visual importante, ausencia de respuesta al tratamiento médico o si tiene lugar un síndrome endocrinológico debido a la hiperproducción de GH (hormona del crecimiento) dando lugar a “Acromegalia o gigantismo” o si tiene lugar la hiperpoducción de cortisol endógeno y con ello la enfermedad de Cushing.
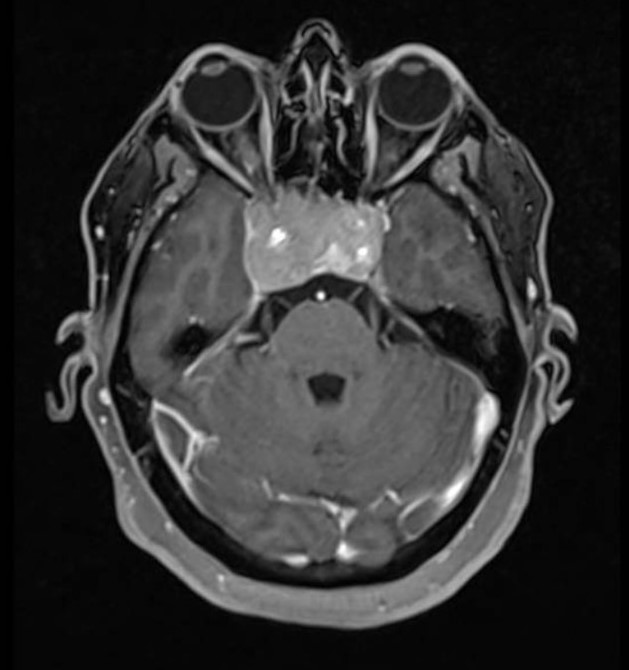
–Hemangioblastomas: Se originan en la fosa posterior siendo el tumor primario más frecuente en adultos en dicha localización. Se trata de un tumor benigno (grado I de la OMS). Suelen darse en adultos jóvenes y el 20% de los casos ocurren como parte de la enfermedad de von Hippel Lindau en cuyo caso pueden presentar también afectación retiniana. Su carácter vascular hace que reciban gran cantidad de flujo sanguíneo. Pueden acompañarse de policitemia. Su tratamiento es quirúrgico.
–Quistes epidermoides: Tienen lugar frecuentemente en la fosa posterior a nivel del ángulo pontocerebeloso, siendo los terceros tumores en frecuencia en dicha localización tras el schwannoma del VIII par craneal y el meningioma. Los epidermoides se componen de tejido escamoso con aspecto perlado perteneciente a células ectodérmicas que quedan atrapadas durante el desarrollo embrionario. La liberación espontánea de su contenido al espacio subaracnoideo puede provocar una meningitis química (meningitis de Mollaret). Presentan un comportamiento benigno, si bien su tendencia al crecimiento lineal hace que precisen tratamiento quirúrgico. Su relación frecuente con arterias vitales, nervios craneales y estructuras troncoencefálicas dificulta la extirpación capsular completa por lo que es frecuente que recidiven y precisen de nueva extirpación quirúrgica transcurridos años de la inicial. No existe lugar para la radioterapia ni la quimioterapia en estos tumores.
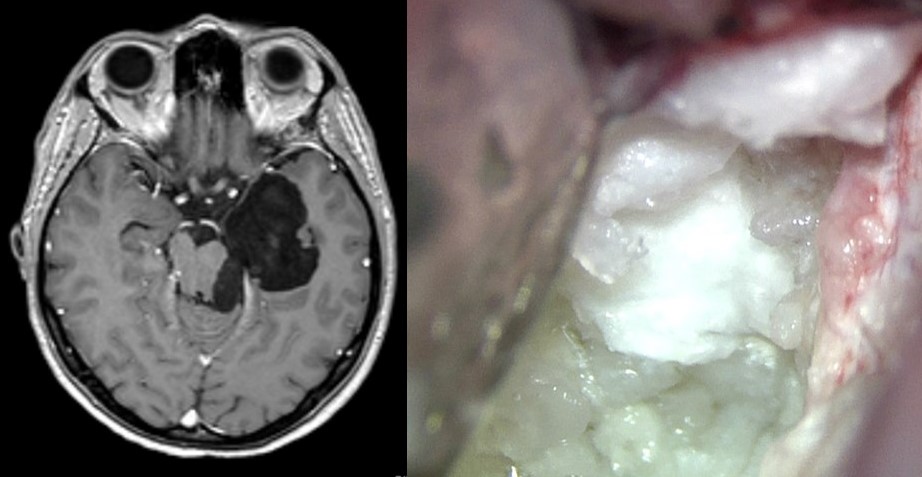
-Linfoma cerebral primario: Existe una creciente incidencia en la aparición del linfoma cerebral primario. Tiene lugar en pacientes adultos, a veces asociado a inmunodepresión congénita o adquirida. Este tumor es conocido como “tumor fantasma” debido a que tiende a desaparecer de las pruebas de imagen si tiene lugar la administración de corticoides. El papel de la cirugía está limitado a la biopsia para confirmación del diagnóstico. El tratamiento del linfoma cerebral primario es la radioterapia y la quimioterapia.
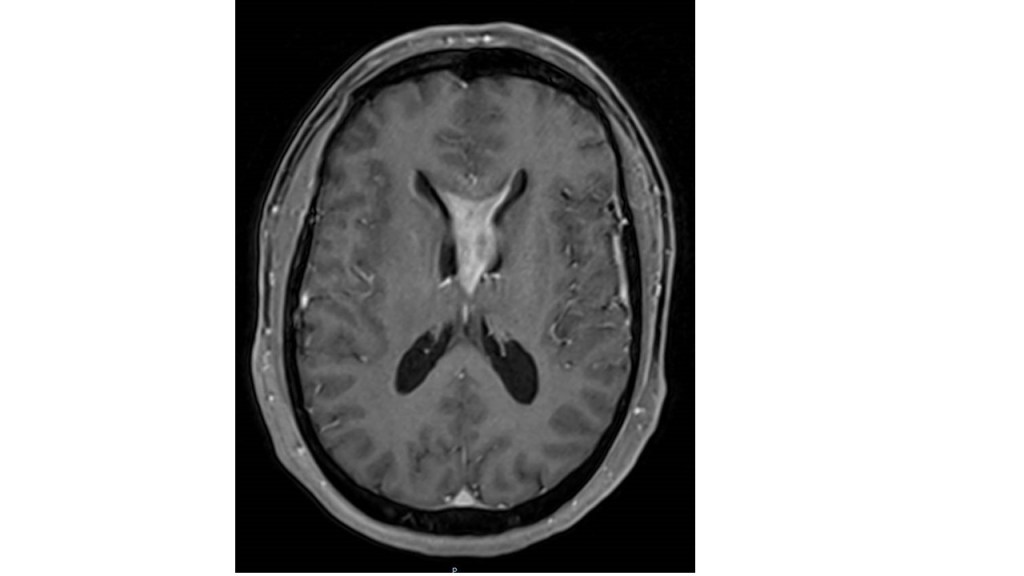
-Metástasis cerebrales: El aumento de los tratamientos para los tumores ha generado un incremento en la tasa de curación y supervencia de los mismos. La aparición de metástasis cerebrales tene lugar sobre todo en pacientes con cáncer de mama y pulmón. Es fundamental llevar a cabo un tratamiento adecuado en cada caso. Gran parte de estos pacientes puede beneficiarse de la cirugía sobre todo aquellos pacientes con metástasis cerebral única en ausencia de afectación tumoral de otros órganos. Tras la intervención se debe administrar tratamiento con radioterapia.
Tumores cerebrales en la edad pediátrica
Los tumores cerebrales representan, después de las neoplasias hematológicas, la segunda causa de cáncer en la edad pediátrica.
Los tumores cerebrales más frecuentes en niños, aparte de los ya citados astrocitomas pilocíticos (grado I de la OMS ) y ependimomas (grado II y III) son los siguientes:
–Meduloblastoma: Tumor originado a partir de células primitivas neuroectodérmicas. Pesenta un alto componente proliferativo (grado IV de la OMS). Tiene lugar en la fosa posterior, característicamente en el techo del cuarto ventrículo, si bien no es rara la localización en hemisferio cerebeloso en adultos jóvenes. Puede dar lugar a la siembra a través del neuroeje, incluso a metástasis fuera del sistema nervioso central. Es frecuente la presencia de hidrocefalia obstructiva en el momento del diagnóstico. Ésta puede precisar una intervención quirúrgica previa a la extirpación del tumor. El tratamiento del meduloblastoma se basa en la extirpación quirúrgica seguida de radio y quimioterapia. Presenta una tasa de curación elevada en los casos en los que no hay diseminación y la cirugía logra eliminar por completo o reducir al mínimo la carga tumoral.
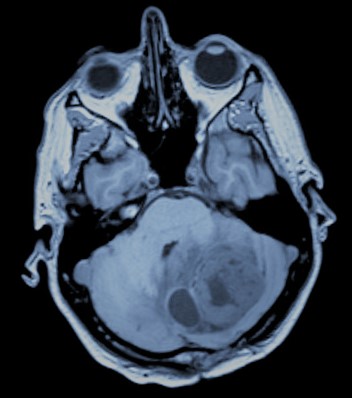
-Craneofaringioma: Típico en niños, aunque puede presentarse también en adultos. Se trata de tumores de origen epitelial derivados de remanentes embrionarios de la bolsa de Rathke, estructura precursora de la adenohipófisis. Ello hace que su localización anatómica tenga lugar en relación al eje hipotálamo-hipofisario. Su extensión a estructuras hipotalámicas da lugar a la aparición frecuente de un síndrome hipotalámico caracterizado por el aumento del apetito con el consiguiente aumento de peso, la diabetes insípida (incapacidad para retener el agua con poliuria y polidipsia), alteración en los ciclos de sueño-vigilia, alteración de la temperatura corporal y de la frecuencia cardíaca. De este modo, pese a exhibir rasgos de tumor benigno su localización profunda, la disfunción endocrinológica que conlleva y su capacidad invasiva con tendencia a la recidiva hacen que su tratamiento sea complejo, teniendo en ocasiones que valorar, además de la cirugía, la radioterapia adyuvante.
-Tumores pineales: Tienen lugar en la glándula pineal o epífisis situada en la parte posterior del diencéfalo y relacionada con la producción de melatonina y con los ciclos de sueño-vigilia. Al igual que sucede con los craneofaringiomas, los tumores pineales aunque típicos en niños, tienen lugar también en adultos. Existen una gran variedad de estirpes de tumores pineales que pueden exhibir grados de agresividad muy variados. En niños los más frecuentes son de tipo germinal (germinomas, sobre todo, y menos frecuentes coriocarcionas, carcinomas embrionarios, tumor de seno endodérmico y teratomas maduros/inmaduros), mientras que en adultos suelen tratarse de gliomas o meningiomas. Estos tumores suelen manifestarse con el llamado Síndrome de Parinaud, una incapacidad para dirigir la mirada hacia arriba en ocasiones acompañada de hidrocefalia obstructiva aguda e hipertensión intracraneal. En estos casos además de llevar a cabo cirugía sobre el tumor puede ser preciso una intervención previa para el tratamiento de la hidrocefalia. La necesidad de tratamiento con quimio o radioterapia está determinado, al igual que el pronóstico, por la estirpe del tumor.
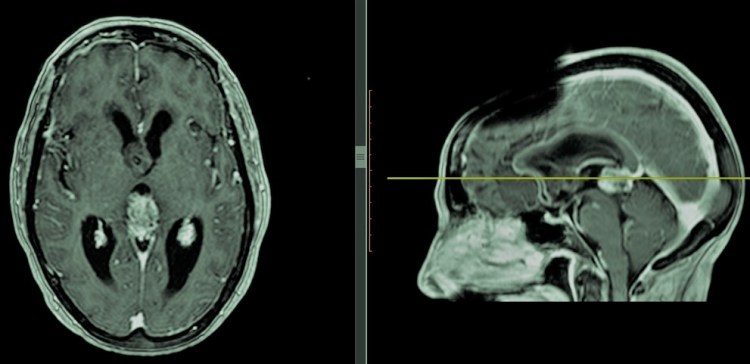
-Tumores del plexo coroideo: Derivados del plexo coroideo, implicado en la producción de líquido cefalorraquídeo a nivel del sistema ventricular, son los tumores cerebrales congénitos más frecuentes. La mayoría de ellos tiene lugar en niños antes de los dos años edad. En su mayoría son benignos (Papiloma de plexo coroideos, grado I de la OMS), si bien existen formas capaces de exhibir mayor agresividad. Su tratamiento es quirúrgico.
-Tumores embrionarios (PNET): Al igual que los meduloblastomas se originan a partir de células primitivas neuroectodérmicas. Su localización sin embargo es supratentorial. Se caracterizan por tener un comportamiento altamente agresivo con posibilidad de diseminación a través del líquido cefalorraquídeo y gran tendencia a la recidiva.







